0. 单细胞多组学揭示肿瘤免疫微环境:系统评述
1. 引言
肿瘤微环境(TME)被定义为一个多层面且动态的生态系统,其核心特征在于深刻的细胞异质性、高度动态性以及复杂的细胞间相互作用。它由肿瘤细胞、免疫细胞、基质细胞、内皮细胞以及非细胞成分如细胞外基质和分泌信号分子组成。当前,免疫检查点阻断(ICB)疗法在癌症治疗中取得了显著的临床效益,这充分证明了靶向TME组成而非仅限于肿瘤细胞的抗癌策略具有巨大潜力。然而,由于ICB疗法目前仅对部分患者有效,深入理解TME中免疫反应的调控机制已成为迫切需求,这将为设计更有效、更精准的抗癌疗法提供指导。
传统批量(bulk)测序方法虽然能够预测新抗原、量化TME中不同细胞类型并分析淋巴细胞受体多样性,但其本质是对细胞群体进行“平均”表征,因此无法充分揭示单个细胞的独特贡献、精细异质性以及复杂的细胞间相互作用。这种局限性阻碍了对癌症生物学和肿瘤免疫学更深层次的理解,并限制了靶向疗法的临床效益。
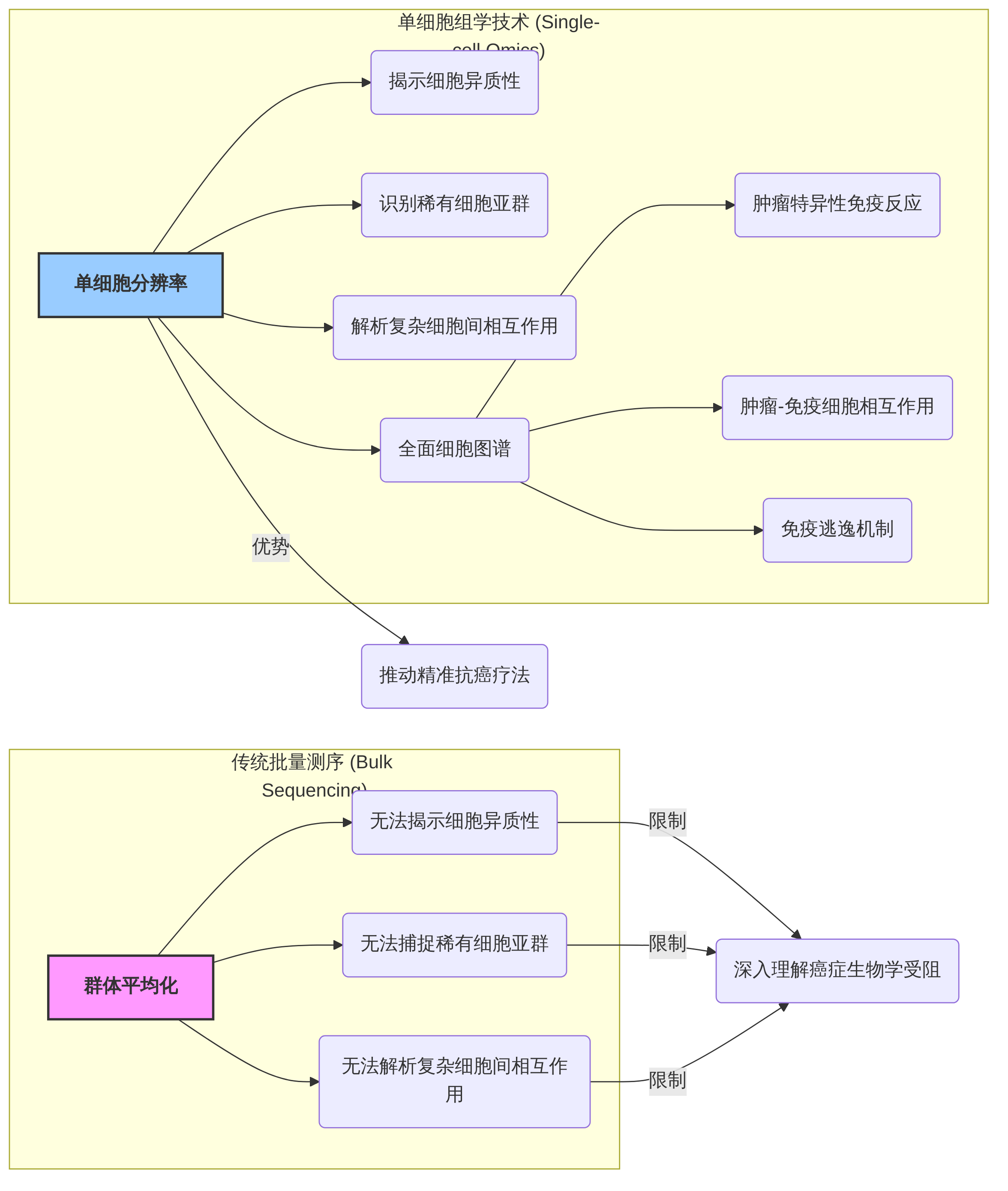
为克服传统方法的不足,单细胞组学技术应运而生并迅速发展,以前所未有的分辨率揭示了TME的细胞和分子复杂性。该技术能够以无偏且全面的方式分析TME的细胞图谱,从而深入剖析肿瘤特异性免疫反应、肿瘤-免疫细胞相互作用以及免疫逃逸的细胞和分子机制。单细胞测序技术在揭示细胞异质性、可塑性、克隆进化以及肿瘤微环境重塑状态方面展现出独特优势,对于预测免疫治疗耐药机制和发现生物标志物至关重要。
进一步地,单细胞多组学技术通过同时整合基因组、转录组、表观基因组、蛋白质组等多种组学数据,彻底改变了分子细胞生物学研究。这些技术在过去十年中通过优化通量和分辨率、多模态整合、独特性和准确性等方面得到了显著改进。结合单细胞测序,多组学方法能够更好地解析癌症生态系统中复杂的细胞状态和表型互动,并探索传统批量多组学方法中被混淆的细胞异质性。随着单细胞转录组学数据集的不断积累以及向单细胞基因组学、表观基因组学和蛋白质组学的进一步扩展,有望不仅解析免疫细胞的细胞组成和功能状态,还将揭示肿瘤和免疫细胞的发育历史、调控网络及细胞间相互作用。更先进的技术甚至已发展到可同时表征单细胞水平的多组学信息和空间信息,这有助于更全面地揭示疾病特异性细胞的表型和功能。
本综述旨在系统性地探讨单细胞多组学技术在TME研究中的应用、技术进展、数据分析挑战及未来展望。通过深入分析这些方法带来的肿瘤细胞异质性和可塑性的更广泛视野,本综述将阐明其如何为合理设计精准免疫肿瘤学治疗提供机会,并为克服靶向药物和检查点阻断剂的当前局限性奠定基础,最终有望为更大比例的癌症患者带来长期临床益处。同时,本综述将明确探讨单细胞技术的潜在缺陷和在癌症治疗中带来的新机遇,旨在为读者提供一个清晰的阅读框架和价值预期。
2. 单细胞多组学技术概览及其在肿瘤免疫微环境研究中的演进
肿瘤是一种高度复杂的生态系统,其内部由多种细胞类型、状态和命运交织而成。肿瘤的发生始于单个细胞,但随着疾病的进展,细胞会不断积累体细胞突变,并分化形成独特的谱系和亚克隆群体,从而产生显著的肿瘤内异质性(Intra-tumoral Heterogeneity, ITH)。这种异质性及其导致的克隆演化在肿瘤免疫逃逸、血管生成和转移等关键生物学过程中扮演着核心角色。传统的批量测序方法由于仅能提供细胞集合的平均信号,无法有效捕捉这种复杂的异质性,从而在临床诊断和患者治疗中造成混淆并构成重大挑战。
为了克服传统方法的局限性,单细胞多组学技术应运而生,并在揭示肿瘤免疫微环境(Tumor Microenvironment, TME)的复杂性方面展现出前所未有的能力。这些技术通过激光捕获显微切割、机器人微操作、荧光激活细胞分选或微流控平台等先进方法实现单细胞分离,并对单个细胞中的多种分子进行条形码标记,从而在单细胞分辨率下对基因组信息进行全基因组表征。相较于传统流式细胞术在可检测标志物数量上的限制,单细胞技术以高分辨率和高颗粒度对细胞类型和状态进行分类,能够深入剖析肿瘤内部及肿瘤间的异质性,并识别出在癌症进展和侵袭中发挥关键作用的稀有细胞亚群,如循环肿瘤细胞(CTCs)、癌症干细胞和经历上皮-间充质转化(EMT)的细胞。
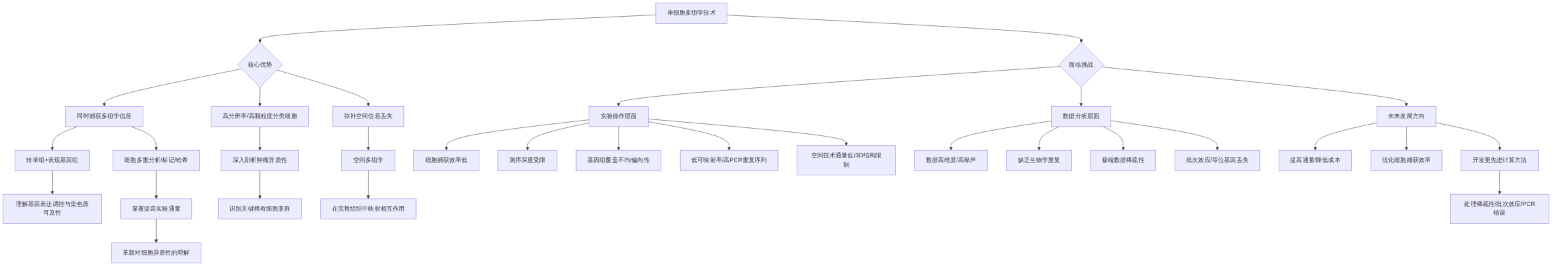
单细胞多组学技术的核心优势在于能够同时捕获不同组学模态的信息,例如将转录组信息与其他组学数据结合,并通过细胞多重分析、细胞标记和细胞哈希等策略显著提高实验通量,从而革命性地提升了对健康与疾病状态下细胞异质性的理解。例如,单细胞转录组测序(scRNA-seq)与单细胞转座酶可及染色质测序(scATAC-seq)的整合应用,能够协同理解基因表达调控与染色质可及性之间的复杂关系。此外,空间多组学技术进一步弥补了传统单细胞方法因细胞解离而导致空间信息丢失的局限性,能够在完整的组织结构中映射单细胞间的相互作用,这对全面理解TME的复杂性至关重要。
尽管单细胞多组学技术潜力巨大,但其在实际应用中仍面临诸多挑战。这些挑战不仅存在于实验操作层面,如细胞捕获效率低、测序深度受限、基因组覆盖不均(由于随机的过度或不足扩增)、对高GC含量区域的强烈偏向性、低可映射率(约20%至30%)以及高水平的聚合酶链反应重复序列等,也延伸至数据分析层面,包括数据高维度、高噪声、缺乏生物学重复以及因低覆盖率导致的极端数据稀疏性、批次效应和等位基因丢失等问题。此外,多数空间技术在处理组织数量方面通量较低,且无法完全捕获组织的3D结构,其捕获面积相对较小,限制了其在完整人类组织切片分析中的大规模应用。
为了应对这些技术瓶颈,新的实验技术和计算分析方法正在迅速发展,以期通过估算缺失信息和开发更稳健的计算工具来克服现有问题。尽管如此,单细胞多组学技术仍需进一步努力以实现完全成熟和标准化。未来的技术发展将着重于提高通量、降低成本、优化细胞捕获效率,并开发更先进的计算方法来有效处理数据稀疏性、批次效应和PCR错误,从而为全面深入揭示肿瘤免疫微环境的复杂性提供更坚实的基础。
2.1 单细胞转录组学(scRNA-seq)
单细胞RNA测序(scRNA-seq)作为当前应用最广泛且已验证其准确性和可扩展性的单细胞测序技术,其技术发展迅速,涵盖液滴微流控与微孔板两大主要平台。早期的scRNA-seq方法(如Smart-seq平台)已迭代至第三代(Smart-seq3),通过整合5'端UMI提升了转录本异构体的量化精度;同时,微流控平台利用珠基系统显著提高了单次样本的细胞检测数量,可分析多达数十万个细胞并检测数千个基因。scRNA-seq对肿瘤异质性、克隆演化和转移扩散的理解产生了深远影响。
scRNA-seq在肿瘤免疫微环境(TME)研究中的主要应用体现在多个方面:首先,它能够高分辨率地重建不同癌症类型中的TME图谱,并结合单细胞表观基因组学,揭示免疫系统的异质性、可塑性和功能多样性。其次,scRNA-seq的无偏性使其能够识别稀有免疫细胞亚群,并分析细胞发育轨迹及细胞状态的动态变化。
在免疫相关因素的探究中,scRNA-seq已被广泛应用于分析多种肿瘤(包括皮肤癌、乳腺癌、肺癌等)的肿瘤内免疫图谱。研究初期主要聚焦于肿瘤浸润淋巴细胞(TILs),特别是细胞毒性CD8+ T细胞。scRNA-seq揭示肿瘤内CD8+ T细胞通常处于功能失调和耗竭状态,表现为效应细胞因子分泌和细胞裂解功能受损,并持续表达T细胞耗竭标志物。通过对数万个细胞的分析,发现CD8+ T细胞从早期效应“过渡”到完全功能失调和耗竭状态是一个连续的细胞谱系,而非离散亚型,表明这些细胞状态是TME刺激下持续激活和分化的结果。
结合肿瘤空间异质性分析,scRNA-seq能够深入理解癌症相关免疫抑制对肿瘤浸润性T细胞功能障碍、免疫抑制性肿瘤T细胞和巨噬细胞的影响机制。例如,对黑色素瘤的scRNA-seq分析揭示了肿瘤和T细胞在空间和功能上的异质性,并展示了肿瘤内T细胞活化、克隆扩增和耗竭程序的多样性。此外,scRNA-seq结合scTCR-seq的应用揭示了PD-1阻断在人体内的作用机制,例如在基底细胞癌和鳞状细胞癌患者中,PD-1阻断后新招募的肿瘤反应性CD8+ T细胞而非预先存在的细胞发挥了主要作用。一项研究还利用scRNA-seq确定了接受检查点抑制剂治疗的黑色素瘤患者的免疫图谱,识别出两种主要的细胞状态:一种是干细胞样记忆CD8 T细胞,主要存在于对治疗有反应的肿瘤中;另一种是功能失调或耗竭的T细胞,常见于耐药肿瘤中,这为免疫治疗响应预测提供了早期探索方向。功能失调的肿瘤反应性CD8+ T细胞尽管效应功能改变,但仍可分泌CXCL13,招募其他免疫细胞到TME,提示其在肿瘤免疫中的潜在新作用。
尽管scRNA-seq在TME研究中展现出巨大潜力,但其仍存在局限性。该技术无法直接提供蛋白或表观遗传信息,且由于单细胞解离效率的差异,目前不适用于TME细胞亚型的精确定量。此外,scRNA-seq数据特有的稀疏性(dropout events)和批次效应是普遍存在的挑战。为缓解这些问题,现有计算方法包括插补算法用于填充缺失值,以及批次校正工具用于消除不同实验批次间非生物学变异。尽管这些方法在一定程度上提高了数据质量和可比性,但其仍存在局限性,例如插补可能引入假阳性,批次校正可能过度校正或无法完全消除复杂批次效应。未来研究方向应侧重于开发更稳健的计算方法,以更准确地处理数据稀疏性和批次效应,并结合多组学技术,全面揭示肿瘤免疫微环境的复杂性。
2.2 单细胞表观基因组学(scATAC-seq, scBS-seq等)
单细胞表观基因组学,尤其是单细胞转座酶可及染色质测序(scATAC-seq)和单细胞亚硫酸氢盐测序(scBS-seq)等技术,通过分析DNA甲基化、染色质状态和染色体组织,为解析肿瘤群体内部显著的细胞间变异性提供了关键工具。这些方法的核心在于揭示基因组特征与细胞功能、命运和身份之间的复杂联系,从而阐明肿瘤进展中的基因组调控机制。
在癌症研究中,表观遗传修饰在不改变DNA序列的前提下调控基因表达,是癌症进展中的重要因素。scBS-seq作为DNA甲基化分析的核心技术,能够揭示细胞亚群内的差异甲基化异质性。例如,在肝细胞癌的研究中,scBS-seq与scRNA-seq的结合应用成功检测到两个亚群中的差异甲基化异质性。当scBS-seq与scDNA-seq结合时,研究者在同一肿瘤中发现了相似的簇集性拷贝数变异(CNVs)模式,这表明基因组结构变异与表观遗传修饰之间可能存在关联。早期的多组学方法如scM&T-seq和scMT-seq已实现DNA甲基化组和转录组的联合分析,而scTrio-seq则进一步扩展到基因组、甲基化组和转录组的三组学分析,为全面理解肿瘤微环境提供了更广阔的视角。
scATAC-seq则主要用于测量顺式调控元件的基因组范围染色质可及性,并识别在单细胞分辨率下驱动功能性染色质状态的反式作用因子。该技术通过利用Tn5转座酶对开放染色质进行片段化并标记衔接子,因其操作简便、耗时少且适用于低细胞数量分析而得到广泛应用。scATAC-seq与scRNA-seq的互补性在于,scATAC-seq能够提供基因调控层面的深刻洞察,补充scRNA-seq提供的基因表达信息,从而更全面地理解肿瘤免疫微环境中细胞命运决定、细胞激活状态和耐药机制。例如,T-ATAC-seq(一种结合转录组和染色质可及性的单细胞分析方法)被用于分析人外周T细胞,揭示了幼稚和记忆CD4+ T细胞状态的反式调控因子,并发现了表面标志物定义的T细胞群体中显著的表观基因组异质性。这种方法对于克隆性T细胞的谱系追踪尤为有用,有助于理解T细胞分化和记忆形成的表观遗传调控机制。值得注意的是,与转录组学不同,耗竭T细胞的表观遗传状态在PD-1阻断后仍保持稳定,这可能是阻碍其持续活化的主要障碍,提示了表观遗传调控在肿瘤免疫治疗中的重要性。
尽管单细胞表观基因组学技术取得了显著进展,但在样本通量、数据覆盖度和检测稀有表观遗传修饰方面仍面临挑战。例如,全基因组亚硫酸氢盐测序(WGBS)虽然覆盖度高,但成本亦高,而简化基因组亚硫酸氢盐测序(RRBS)则在覆盖范围和成本之间取得平衡。新兴技术如单细胞组合索引(single-cell combinatorial indexing)的引入,如单细胞组合索引Hi-C(single-cell combinatorial indexed Hi-C)以及组合细胞索引策略下的scATAC-seq,使得在高通量下对异质细胞群中的开放染色质区域和高阶染色质差异进行鉴定成为可能,提高了效率和数据质量,并被用于关联核型和细胞周期状态的差异。此外,Paired-Tag和CoTECH等技术通过组合条形码,实现了转录组和染色质占据的高通量联合检测,为理解基因调控机制提供了更全面的视角。组蛋白修饰分析则利用CUT&Tag等方法,通过抗体导向的转座酶标记,实现单细胞水平的表观基因组图谱绘制。这些技术的不断发展将有助于克服当前限制,推动对肿瘤免疫微环境复杂调控机制的深入理解。
2.3 单细胞蛋白质组学(CITE-seq, sc-WGS等)
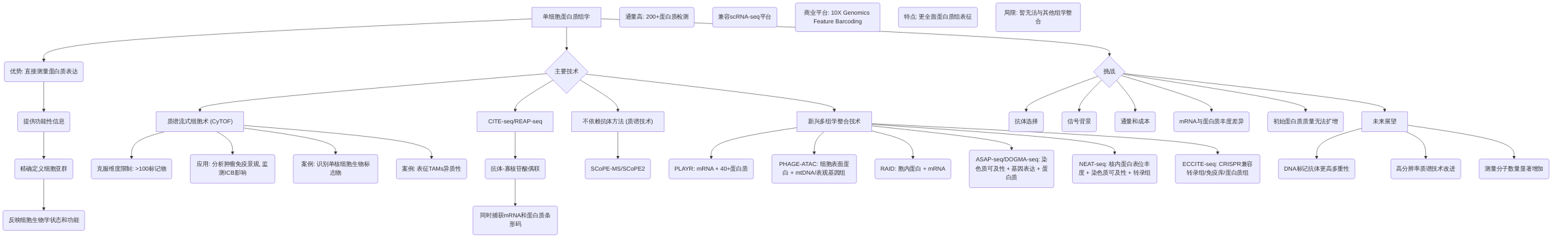
单细胞蛋白质组学技术在揭示肿瘤免疫微环境(TME)的复杂性方面展现出独特的优势,因其能够直接测量蛋白质表达,从而提供比核酸组学更直接的功能性信息,例如识别细胞表面标志物以精确定义细胞亚群,并反映细胞的生物学状态和功能 。尽管信使RNA(mRNA)和蛋白质丰度之间存在差异,且初始蛋白质质量无法扩增给单细胞蛋白质组学带来了挑战,但研究主要集中于开发更灵敏的蛋白质检测方法 。
早期方法如PLAYR已能在单细胞中同时量化40多种mRNA和蛋白质 。质谱流式细胞术(CyTOF)的引入克服了传统流式细胞术的维度限制,通过使用稀有元素同位素偶联单克隆抗体,理论上可检测的标记物数量可达100个以上,实际应用中已能达到45个标记物,从而准确确定细胞的功能特性 。CyTOF在分析新鲜和冷冻的外周血单核细胞(PBMC)或肿瘤组织单细胞方面展现出强大的能力和准确性 。该技术已被广泛应用于分析肿瘤免疫景观,监测免疫检查点阻断(ICB)如何影响免疫细胞群体及其在响应者和非响应者中的功能 。例如,CyTOF已被用于表征黑色素瘤患者在抗PD-1免疫疗法前后免疫细胞的功能状态,发现PBMCs中 单核细胞的频率可作为区分治疗反应者和非反应者的生物标志物 。此外,CyTOF在识别TME中髓系细胞群体方面也发挥作用,例如通过分析肺腺癌和非癌性肺组织中的肿瘤相关巨噬细胞(TAMs)的异质性表达谱,并发现其与不良临床预后相关 。
为了克服CyTOF在检测维度上的局限性,CITE-seq(Cellular Indexing of Transcriptomes and Epitopes by Sequencing)于2017年出现,通过将抗体与寡核苷酸偶联,并利用液滴微流控系统同时捕获mRNA和细胞表面蛋白质条形码,极大地提高了转录组和蛋白质组联合分析的通量,兼容多种单细胞RNA测序(scRNA-seq)平台 。CITE-seq通过DNA标记抗体技术,使得抗体标记蛋白的检测数量显著增加到200多个,是CyTOF检测数量的五倍 。REAP-seq与CITE-seq类似,通过不同抗体-DNA条形码偶联方式实现mRNA和表面蛋白的量化 。商业平台如10X Genomics的Feature Barcoding进一步改进了条形码策略,实现了转录组和蛋白质组的同步分析 。
单细胞蛋白质组学技术还包括不依赖抗体的方法,如SCoPE-MS和SCoPE2,它们利用质谱技术实现更全面的蛋白质组表征,但目前尚无法与其他组学整合 。新兴技术如PHAGE-ATAC和RAID分别实现了细胞表面蛋白与线粒体DNA/表观基因组修饰的联合分析以及胞内蛋白(包括磷酸化蛋白)和mRNA的联合量化 。ASAP-seq和DOGMA-seq则在单细胞中同时分析染色质可及性、基因表达和蛋白质水平 。NEAT-seq进一步实现了核内蛋白表位丰度、染色质可及性和转录组的三重分析 。ECCITE-seq扩展了蛋白质多组学领域,整合了CRISPR兼容的转录组、免疫库和蛋白质组索引,包括克隆型分析和CRISPR介导的扰动,提供了高灵敏度和单细胞分辨率的分析能力 。
尽管单细胞蛋白质组学取得了显著进展,但在抗体选择、信号背景、以及通量和成本方面仍面临挑战 。目前,大多数基于抗体或微流控的单细胞蛋白质组学技术,可以对每个样本中数十万个细胞的最多50种蛋白质进行检测 。未来,随着DNA标记抗体技术在更高多重性方面的进一步发展以及高分辨率质谱技术的改进,测量到的分子数量有望显著增加 。
2.4 多组学整合技术(Multi-omics Integration Technologies)
新兴的多组学整合技术通过同时测量多个组学层面的信息,显著克服了单一组学技术的局限性,从而提供了对细胞生物学更为全面的理解。这些技术能够揭示基因表达与染色质可及性、基因表达与蛋白质表达之间等不同组学层面间的协同作用与复杂关联性,例如基因表达、表观遗传修饰和蛋白质丰度之间的相互关系,进而更全面地阐明肿瘤免疫微环境(TME)中的细胞状态转换、信号通路激活以及细胞间相互作用的调控机制。
在技术发展方面,多种创新方法应运而生。例如,CITE-seq和REAP-seq技术能够同时测量单细胞中的转录组和蛋白质组,改进了免疫细胞表型的表征并成功实现了自然杀伤细胞的亚聚类。SHARE-seq则实现了转录组和表观基因组的同步测量,并识别了染色质可及性在转录组调控中的启动作用,有助于推断细胞分化。更进一步的三组学技术,如scTrio-Seq,能够结合并分析单细胞的基因组拷贝数变异、DNA甲基化和基因表达,已被成功应用于识别肝细胞癌中具有不同恶性潜力的异质亚群。此外,scCOOL-Seq同时分析染色质状态、DNA甲基化和拷贝数变异(CNVs),揭示了父系和母系等位基因之间DNA甲基化的差异。scNMT-Seq则联合分析核小体占据、DNA甲基化和mRNA转录,并在胚胎干细胞分化过程中发现了所有三个分子层面在发育相关区域独特的动态偶联。其他多组学方法,如G&T-Seq、DR-Seq、scMT-Seq、scM&T-Seq、Simultaneous multiplexed profiling of RNA and protein、scNOMe-Seq、SIDR-Seq和sciCAR也为细胞生物学的多层面理解提供了工具。
将空间信息与分子图谱相结合,进一步完善了细胞身份的定义,这对于理解免疫系统细胞之间的相互作用以及肿瘤与其他TME细胞类型之间的串扰至关重要。空间转录组学技术如seqFISH+能够以亚细胞分辨率对组织中的转录本进行超分辨成像,可成像超过10000个基因。通过将细胞条形码策略应用于单细胞RNA测序(scRNA-seq),可以对组织中的局部细胞进行原位条形码标记,并通过解复用物理位置与检测到的条形码序列来恢复空间信息。例如,Slide-seq和HDST利用阵列式条形码珠粒捕获空间全转录组,重建的空间图谱分辨率取决于设计的珠粒阵列。这些空间信息对于理解肿瘤发生和免疫逃逸至关重要,例如TCR空间异质性反映了基因组内的肿瘤异质性,并且肿瘤内PD-L1+巨噬细胞的空间分布影响了T细胞浸润。
尽管多组学整合技术取得了显著进展,但在数据处理和分析方面仍面临挑战。匹配的多组学实验方法(如10x Multiome、SHARE-seq、DBiT-seq、SNARE-seq、CITE-seq等)虽然在计算上更容易处理,因为不同的模态可以绑定到单个细胞或单个位点,但每个模态的测序能力可能会导致整体样本量减少。此外,数据异质性、数据对齐以及信息丢失或偏差是当前多组学整合技术面临的主要挑战。现有的计算方法,包括基于图嵌入和深度学习的方法,在实现无缝整合方面取得了一定进展,但仍存在局限性。未来的研究需要开发更鲁棒和可解释的整合框架,以应对这些挑战,从而更准确地识别新的生物标志物和治疗靶点。
3. 单细胞多组学在肿瘤免疫微环境中的应用
单细胞多组学技术通过提供前所未有的细胞和分子分辨率,极大地革新了我们对肿瘤免疫微环境(TME)复杂性的理解,并为肿瘤免疫治疗的精准化提供了新的视角 。这些技术不仅能够精细描绘TME中各类细胞的表型与转录程序,还能深入揭示肿瘤免疫逃逸的机制并识别潜在的治疗靶点 。例如,单细胞RNA测序(scRNA-seq)与质谱细胞计数(CyTOF)等方法结合使用,能够确认新发现的细胞表型,并揭示T细胞在TME中的分化轨迹、克隆扩增及其对PD-1阻断治疗的反应,这些是单一组学技术难以实现的突破性发现 。

当前,单细胞多组学在TME研究中的应用主要聚焦于系统性地解析其异质性、细胞间通讯网络、肿瘤演进与耐药机制以及免疫治疗响应的预测与生物标志物的发现。在多组学整合策略方面,研究人员正探索将不同模态的数据(如基因组、转录组、蛋白质组和空间信息)进行有效融合,以克服数据量大、异质性高和数据稀疏性等技术挑战,从而构建对TME更为全面的认识 。尽管目前在整合不同类型数据方面尚未形成完全共识,但通过纵向样本的多组学数据分析,有望识别出更具信息量的动态生物标志物,这对于合理设计临床试验和精准治疗至关重要 。
3.1 肿瘤免疫细胞亚群的精细解析
单细胞多组学技术,如单细胞RNA测序(scRNA-seq)和质谱细胞计数(CyTOF),以前所未有的分辨率揭示了肿瘤免疫微环境(TME)中免疫细胞的精细亚群及其复杂功能,突破了传统基于少量标志物分类的局限性,并揭示了免疫细胞状态和功能的连续性与可塑性 。
在TME中,肿瘤浸润淋巴细胞(TILs)和肿瘤浸润髓系细胞(TIMs)的精细解析尤为关键 。研究发现,肿瘤内的细胞毒性CD8+ T细胞主要处于功能失调和耗竭状态,其TCR序列和转录组特征可进一步区分肿瘤反应性与旁观者T细胞,这对于理解肿瘤免疫逃逸机制至关重要 。此外,scRNA-seq在乳腺癌中识别了T细胞的连续细胞状态,并提示TCR信号和环境刺激均可调节T细胞功能,从而将TCR克隆型与T细胞表型紧密结合 。CyTOF在透明细胞肾癌(ccRCC)中识别出PD-1+耗竭T细胞的CD38多态性表达,进一步细化了耗竭T细胞的特征 。
CD4+辅助T细胞在TME中也表现出显著的异质性和功能可塑性,包括促炎性T辅助细胞和免疫抑制性调节性T细胞 。例如,CXCL13+ TH1样细胞在微卫星不稳定型结直肠癌中富集,并预示对免疫检查点抑制剂(ICB)治疗的更高响应,这表明特定CD4+ T细胞亚群可能作为治疗反应的生物标志物 。调节性T细胞(Treg)在肿瘤免疫抑制中发挥关键作用,其在TME中的克隆扩增和激活状态与不良预后密切相关 。
单细胞测序技术还深入揭示了髓系细胞群体的复杂性。树突状细胞(DCs)包括cDC1、cDC2和LAMP3+活化DC亚群,它们在肿瘤抗原提呈、T细胞启动和调节T细胞功能方面发挥多重抗肿瘤作用 。通过RNA Velocity分析和基于线粒体的谱系追踪,研究发现一部分LAMP3+树突状细胞可能从肿瘤迁移到肝脏淋巴结,从而触发系统性适应性免疫反应,揭示了免疫细胞在不同组织间的动态迁移过程 。
肿瘤相关巨噬细胞(TAMs)表现出高度异质性,其功能角色在不同癌症类型和阶段中存在争议,且与经典的M1/M2极化范式不完全吻合 。单细胞技术识别出与不良预后相关的TAM亚群,并通过对其进行特定抑制剂处理,揭示其在肿瘤进展中的作用 。例如,CyTOF在肺腺癌中揭示了PPARγ表达的肿瘤富集巨噬细胞可能导致TME免疫抑制 。在ccRCC中,还识别出一种与免疫抑制T细胞亚群高度相关的特殊CD38+ TAM亚群,且发现多种TAM亚群的丰度可预测患者的无进展生存期 。这些发现突破了传统分类的局限,揭示了TAMs在肿瘤免疫逃逸中的复杂作用。
综上所述,单细胞多组学技术通过精细解析TME中免疫细胞的亚群特征和功能,极大地深化了我们对肿瘤进展和免疫逃逸机制的理解。未来的研究应进一步结合多组学数据,探索免疫细胞亚群间的相互作用及其与肿瘤细胞、基质细胞的复杂网络,为开发更精准的肿瘤免疫治疗策略提供理论依据。
3.2 肿瘤细胞和基质细胞的异质性及其对免疫的影响
单细胞多组学技术已成为解析肿瘤细胞和基质细胞内在异质性及其对肿瘤免疫微环境(TME)影响的关键工具。肿瘤细胞的异质性不仅体现在遗传层面,还涉及其表型可塑性、干细胞样特征及克隆演进,这些因素共同塑造了肿瘤的进展和对治疗的响应。
单细胞DNA测序(scDNA-Seq)在揭示肿瘤内异质性和克隆演化方面发挥了重要作用。例如,针对三阴性乳腺癌的研究通过scDNA-Seq检测单个细胞的非整倍性演化,发现存在间断性拷贝数变异(CNVs)演化,继而出现稳定的克隆扩增。另一项研究则追踪了接受化疗的三阴性乳腺癌患者单细胞的基因组演化,发现两个不同的亚克隆群体呈现出灭绝与持续并存的现象。新开发的Slide-DNA-seq空间技术能够在保留局部肿瘤结构的同时,发现不同的肿瘤克隆及其CNVs,揭示了空间上不同的克隆群体以及与克隆变异和局部TME相关的基因。这些研究强调了仅靠遗传特征无法完全概括肿瘤细胞多样性与动态性的局限性,并提示整合其他组学数据对于揭示肿瘤细胞表型在进展和治疗过程中演变的重要性。
肿瘤细胞的干细胞样特征和高可塑性细胞状态(HPCS)是肿瘤进展和化疗耐药的关键驱动因素。scRNA-seq对少突胶质瘤患者癌细胞的分析揭示了部分未分化恶性细胞具有干细胞表型和增殖潜力,提示癌症干细胞在癌症演进中的核心作用。此外,结合基因工程小鼠模型和scRNA-seq的研究发现,高可塑性细胞状态的TIGIT+细胞被标记为促肿瘤进展和化疗耐药的转化肿瘤细胞。胶质母细胞瘤的scRNA-seq研究也表征了四种具有特定分子特征的恶性细胞亚群,并通过细胞条形码和谱系追踪,证实了恶性细胞在不同亚群之间的可塑性。这些研究进一步揭示了肿瘤细胞的基因组变异、转录组重编程和表型可塑性对TME免疫景观的深远影响。
肿瘤细胞通过多种机制直接影响免疫细胞的功能和浸润。它们持续受到宿主免疫系统的选择压力,通过“免疫编辑”获得免疫逃逸的体细胞突变。例如,scRNA-seq分析在接受免疫检查点阻断(ICB)治疗的黑色素瘤患者中识别出一种与ICB耐药相关的肿瘤细胞程序,即CDK4/6通路上调,该程序导致T细胞排除和免疫逃逸。此外,头颈癌(HNSCC)恶性细胞中表达的部分上皮-间充质转化(p-EMT)程序驱动了有效转移。肿瘤来源的趋化因子,如CXCL9/10的表观遗传沉默介导了T细胞在小鼠卵巢癌模型中的排除,而CXCL1的分泌则在小鼠胰腺导管腺癌(PDAC)模型中招募髓系细胞并耗竭T细胞,这些都是调节TME的肿瘤内在机制。通过分析RNA测序数据与全外显子组或全基因组测序数据结合,可以预测患者特异性的癌症新抗原,这有望引发抗癌免疫反应。
除了肿瘤细胞,TME中的非恶性基质细胞,如癌相关成纤维细胞(CAFs)和内皮细胞,也引入了显著的细胞异质性,它们的作用可以支持或对抗肿瘤进展。成纤维细胞在肿瘤-免疫细胞串扰中发挥积极作用,某些CAF亚群与癌症细胞增殖、侵袭增加、药物抵抗以及抗肿瘤免疫力降低相关。
未来研究应继续利用单细胞多组学技术,深入解析肿瘤细胞和基质细胞的复杂互作网络,以期发现更多新型预测生物标志物和理解肿瘤内在免疫逃逸机制。例如,进一步优化新抗原的精准性和时间效率以实现临床转化,以及探索更多整合组学数据的方法,以克服当前精准医疗的局限性。
3.3 细胞状态与功能异质性解析及复杂调控网络
单细胞多组学技术为深入解析肿瘤免疫微环境(TME)中细胞的功能状态和动态变化提供了前所未有的分辨率。其中,单细胞RNA测序(scRNA-seq)和质谱流式细胞术(CyTOF)生成的数据,通过无监督聚类、细胞分类、伪时间排序等计算分析方法,能够有效揭示TME中细胞功能状态的动态变化和异质性 。例如,单细胞转录组学已识别出TME中多种免疫细胞谱系(如T细胞和巨噬细胞)存在持续的激活和分化轨迹,这凸显了微环境与免疫细胞之间复杂的串扰,从而调节其分化和功能 。计算算法如Monocle/Monocle 2能够将细胞投射到分化轨迹上,构建“伪时间”轴,而RNA velocity则基于剪接和未剪接转录本的比例预测细胞的未来状态 。此外,结合单细胞CyTOF和scRNA-seq分析早期肺腺癌的肿瘤细胞、邻近正常组织和血液样本,揭示了肿瘤浸润性髓系细胞可能损害抗肿瘤免疫 。另一项研究通过同时进行scRNA-seq和CyTOF,揭示了免疫检查点治疗期间淋巴和髓系肿瘤内区室的显著重塑 。
尽管单细胞转录组学在揭示细胞状态方面取得了显著进展,但其预测的分化轨迹纯粹是表型上的,不一定反映细胞谱系之间的真实遗传关系 。此外,scRNA-seq衍生的图谱中缺少细胞身份的几个关键方面,包括表观遗传状态、蛋白质谱和空间位置 。因此,从scRNA-seq的“快照”式数据转向多模态测量和整合基因组、表观基因组、转录组、蛋白质组和空间组织数据集,将极大地扩展单细胞组学在免疫学研究中的能力,并最终构建基因调控网络,从而全面阐明复杂生物系统中纯细胞群体的表型和行为 。
多组学整合在揭示复杂调控网络方面具有独特优势。例如,结合成像质谱流式细胞术(IMC)测量转录组和蛋白质组标记,发现在群体水平上HER2 mRNA和蛋白表达具有良好的相关性,但在单细胞水平上则不尽然,这强调了单细胞分辨率在理解细胞状态异质性方面的必要性 。同时分析基因表达、表观遗传修饰和蛋白质丰度,能够更全面地理解肿瘤免疫微环境中的细胞状态转换、信号通路激活以及细胞间相互作用的调控机制 。TME的异质性还体现在细胞间的相互作用,这种相互作用受到其表达/分泌的分子、携带的抗原或受体多样性的影响 。致癌通路与耐药性以及TME中免疫系统的调节密切相关 。通过解析TME的不同异质性层面,可以获得机制性依据,从而释放免疫检查点阻断剂和靶向药物的协同潜力 。表观遗传状态,如染色质可及性,在调控T细胞命运和功能中扮演关键角色,其在解释耗竭T细胞对PD-1阻断响应不持久的原因方面具有重要意义。例如,PD-1阻断在某些耗竭T细胞中无法恢复其完全功能,可能与染色质重塑等表观遗传障碍有关,这些障碍阻止了细胞重编程到更有效的状态。理解这些复杂的表观遗传调控网络,对于开发更有效的免疫疗法至关重要。
3.4 细胞间相互作用网络构建
单细胞多组学技术在解析肿瘤免疫微环境(TME)中的复杂细胞间通讯网络方面发挥着关键作用,这对于理解肿瘤进展和免疫治疗反应至关重要。研究表明,TME内的细胞类型并非孤立存在,而是通过分泌分子、细胞接触等方式形成精密的相互作用网络。
在TME中,关键的细胞间相互作用模式多样且复杂。例如,树突状细胞(DCs)作为抗原提呈细胞,通过分泌趋化因子、细胞因子和共刺激分子来启动和调节T细胞功能。与此同时,DCs的功能也受到自然杀伤(NK)细胞和调节性T细胞(Treg细胞)的强烈调节,这种多向互动揭示了免疫细胞间复杂的调控环路。此外,肿瘤相关巨噬细胞(TAMs)也表现出与微环境内其他不同细胞类型相互作用的能力,这些相互作用共同驱动肿瘤的进展和免疫逃逸。
不同类型的细胞间相互作用在肿瘤生物学中扮演着不同的角色。例如,某些相互作用可能促进免疫抑制通路,从而帮助肿瘤逃避免疫监视;而另一些则可能激活促瘤通路,直接支持肿瘤生长和转移。以胰腺导管腺癌为例,整合单细胞RNA测序(scRNA-seq)和空间转录组数据发现,应激反应癌细胞与释放IL-6的炎症成纤维细胞共定位,支持了IL-6诱导的癌症应激反应机制,这是一种典型的促瘤相互作用模式。
在识别这些信号通路时,不同的推断工具各有优缺点。尽管传统的免疫组织化学(IHC)和免疫荧光(IF)技术能够揭示TME的细胞结构,但其可检测标记物数量有限。新兴的基于抗体的成像技术,如成像质谱流式细胞术(IMC)和多重离子束成像(MIBI),通过定量多达40种标记物,能够生成类似组学的数据,显著提升了识别细胞间相互作用的深度和广度。例如,成像CyTOF已被用于分析乳腺肿瘤样本的亚细胞分子图谱,揭示了局部肿瘤生态系统的基因组调控、细胞组成和细胞邻域,及其在乳腺癌预后中的临床预测作用。
多组学整合显著提升了通讯推断的可靠性。Schulz及其同事通过改造IMC,实现了在同一细胞上同时测量转录组和蛋白质组标记物,并揭示了HER2 mRNA和蛋白质表达在细胞群体水平上相关,但在单细胞水平上不相关的现象。他们还展示了CXCL10(一种促进T细胞招募的趋化因子)表达细胞的聚集模式,这突显了空间模式对于理解组织生物学的重要性。此外,利用批量蛋白质组学和转录组学数据重建卵巢腺癌细胞间通讯图谱的案例也表明,通过整合不同层面的组学数据,可以更全面地理解TME中细胞间配体-受体相互作用和细胞通讯网络,并评估其对肿瘤进展和免疫逃逸的影响。这些技术的进步共同推动了对TME复杂性的深入理解,为精准免疫肿瘤学铺平了道路。
3.5 肿瘤免疫治疗响应预测与生物标志物发现
单细胞多组学技术在精准免疫肿瘤学中展现出巨大潜力,特别是在预测患者对免疫检查点抑制剂(ICI)或其他免疫疗法的响应、识别耐药机制以及发现新型治疗靶点和生物标志物方面。通过分析治疗前后肿瘤微环境(TME)的动态变化,单细胞多组学能够揭示与治疗响应或耐药机制相关的特定细胞亚群或基因表达特征。尽管肿瘤突变负荷(TMB)、微卫星不稳定性以及PD-L1表达已与对PD-1/PD-L1轴阻断的反应相关联,但目前仍缺乏能够明确区分响应者和非响应者的稳健生物标志物。
单细胞研究已识别出多种与免疫治疗响应相关的特定细胞亚群。例如,在小鼠模型中发现的“祖细胞样”肿瘤内CD8+ T细胞亚群(Tcf1+),在PD-1阻断或TCR刺激后表现出增殖和分化为细胞毒性“终末耗竭”CD8+ T细胞的多功能性,提示其可能对ICI治疗的持久响应有贡献。在人类癌症TME中也观察到TCF1+ TILs和CXCR5+ TILs的存在,其与小鼠对应物的相似性仍在探讨中。此外,激活的树突状细胞(DC)亚群和/或增殖性肿瘤内T细胞亚群的存在可能与免疫治疗的临床结果呈正相关。PD-L1阻断通过激活或恢复DC功能增强T细胞启动,从而产生有效的抗癌T细胞免疫,且DC基因特征与肾癌和非小细胞肺癌(NSCLC)患者接受PD-L1阻断治疗后的总生存期改善相关。
单细胞技术在预测免疫治疗耐药性方面也发挥着重要作用。scRNA-Seq通过揭示与肿瘤浸润性T细胞减少相关的基因表达程序,为预测免疫疗法耐药性提供了潜在方法,从而指导潜在靶点以延迟或对抗耐药性。例如,对免疫治疗耐药的黑色素瘤高分辨率图谱的绘制,揭示了耐药肿瘤中功能失调或耗竭的T细胞更为常见,而响应肿瘤则富集了干细胞样记忆CD8+ T细胞,这为治疗前预测耐药性提供了潜在依据。
多组学整合在预测免疫治疗反应和发现生物标志物方面具有显著优势。单细胞CyTOF结合scRNA-Seq分析早期肺腺癌TME的研究表明,肿瘤浸润性髓系细胞可能损害了抗肿瘤免疫,提示髓系细胞在TME中的关键调控作用。此外,同期scRNA-Seq和CyTOF被用于揭示免疫检查点治疗期间淋巴和髓系肿瘤内微环境的显著重塑。这些发现对精准免疫肿瘤学具有重要启示,有助于识别治疗前后外周血或肿瘤内的特定免疫细胞亚群或基因表达程序作为预测性生物标志物。例如,治疗前外周血中CD14+CD16-HLA-DR+单核细胞的频率与黑色素瘤患者对抗PD-1免疫治疗的反应高度相关,可用于患者分层。
在CAR-T细胞疗法中,单细胞多组学也为预测治疗反应、不良事件和神经毒性提供了生物标志物。Deng等人的研究深入探讨了输注产品(IPs)中CAR-T细胞的转录表型与弥漫性大B细胞淋巴瘤患者临床结果的关系,揭示了IPs中记忆表型CAR-T细胞的富集预示着积极的临床反应,而耗竭表型CAR-T细胞的富集则与疾病进展相关。他们还识别出IPs中一部分单核细胞样细胞与高级别免疫效应细胞相关神经毒性综合征(ICANS)显著相关。Sheih等人通过配对scRNA-seq和scTCR-seq,识别出在及时增加相对频率(IRF)克隆型中的CD8+ CAR-T细胞高表达T细胞细胞毒性和增殖的基因特征,提示其在抗肿瘤反应中的有效作用。这些研究强调了单细胞数据在指导个体化治疗方案制定中的潜力。
尽管单细胞多组学在预测免疫治疗反应方面前景广阔,但其临床转化仍面临障碍。目前研究多受限于样本量较小,且公共数据的可用性限制了预测模型的优化和验证。未来研究需扩大样本量,并开发更完善的多组学数据整合与分析方法,以充分发挥单细胞多组学在精准免疫肿瘤学中的潜力,促进新型生物标志物的发现和临床应用。
3.6 肿瘤演进与耐药机制的解析
单细胞多组学技术在深入解析肿瘤细胞自身的演变和对治疗的耐药性方面展现出独特的优势。肿瘤细胞在肿瘤发生、转移和药物抵抗过程中表现出显著的异质性、动态变化和克隆演进。通过单细胞DNA测序,研究人员得以追踪肿瘤内异质性和克隆演化,例如在三阴性乳腺癌中观察到的断裂拷贝数演化以及化疗后亚克隆群体的持续性与灭绝。
单细胞RNA测序(scRNA-seq)在理解肿瘤内异质性、克隆演化和转移扩散方面产生了重大影响。以黑色素瘤为例,通过scRNA-seq分析免疫检查点阻断(ICB)治疗前后的患者样本,Jerby-Arnon等发现ICB耐药程序,即黑色素瘤肿瘤细胞中CDK4/6通路的显著上调,该程序导致T细胞排除和免疫逃逸。这项研究进一步揭示,ICB治疗塑造了肿瘤细胞的表达谱,使其趋向于耐药程序,提示了恢复的抗肿瘤免疫力驱动下的肿瘤细胞免疫编辑作用。在乳腺癌中,对多西他赛耐药的MCF7乳腺癌细胞的单细胞分析揭示了具有干细胞样表型的细胞亚群,并识别出LEF1是药物抵抗中的关键分子调控因子。
单细胞多组学技术为识别肿瘤细胞的高可塑性细胞状态(HPCS)、转移相关基因特征以及耐药性关键分子调节因子提供了深入见解。例如,scTrio-Seq能够结合基因组拷贝数变异、DNA甲基化和基因表达,识别肝细胞癌中具有不同恶性潜力的异质亚群,从而揭示肿瘤演进的机制。在胶质母细胞瘤药物抵抗中,单细胞谱系分析揭示了遗传和表观遗传的相互作用。慢性淋巴细胞白血病(CLL)的研究结合单细胞转录组和DNA甲基化组数据,构建了谱系树,发现某些CLL谱系对药物治疗更敏感,并揭示了受药物治疗影响的细胞差异表达与特定细胞信号通路相关的基因。
此外,单细胞多组学技术在阐明肿瘤免疫逃逸机制方面发挥关键作用。肿瘤细胞的免疫逃逸机制包括抗原提呈机制的破坏(如HLA杂合性缺失或B2M功能丧失突变)、新抗原耗竭(通过DNA拷贝数丢失、转录抑制或表观遗传抑制选择)以及IFN和IL-2信号通路组分的突变。多模式Perturb-CITE-seq方法通过整合RNA和蛋白质谱分析与Cas9基因组敲除筛选,验证了已知的免疫检查点抑制剂(ICI)耐药机制,并揭示了新的CD58相关耐药机制。具体而言,该方法发现下调CD58表达可诱导恶性细胞上PD-L1的表达,并减少CD8+ T细胞上CD58-CD2轴的共刺激信号。这些发现为理解肿瘤免疫逃逸机制和开发联合疗法提供了新的靶点。例如,靶向免疫逃逸程序中CDK4/6的信号激活可以抑制药物抵抗程序并增强ICI疗效。表观遗传机制在调节免疫细胞分化和功能中也扮演着关键角色,耗竭性T细胞的表观遗传状态在PD-1阻断后仍然稳定,这可能是阻止其持续活化的主要障碍,预示着功能障碍的程度可能进一步受到表观遗传状态的影响,进而影响耗竭性T细胞对ICB治疗的再激活能力。
4. 单细胞多组学数据分析挑战与机遇
单细胞多组学技术的飞速发展为深入剖析肿瘤免疫微环境(TME)的复杂性提供了前所未有的机遇,但同时也带来了显著的计算和数据分析挑战。

这些挑战主要体现在数据本身的复杂性、技术局限性以及不同组学数据类型间的整合难题。当前单细胞多组学数据分析面临的核心问题包括数据稀疏性、高维度、批次效应、不同平台间数据整合的复杂性、低映射率、受限的捕获率以及高水平的PCR重复序列等。
在数据固有特性方面,单细胞数据普遍存在高维度和稀疏性问题,即每个细胞的测量特征数量庞大,但许多特征的表达量为零,这给下游的统计分析和机器学习模型带来了巨大挑战。例如,scRNA-seq技术捕获转录本的效率有限,导致数据丢失率高且噪声水平高于传统的批量RNA-seq,特别是3'端转录本捕获策略可能导致大量非信息性转录本被测序,进而造成测序成本的浪费并限制了对特定感兴趣转录本的有效检查。
技术层面,单细胞测序技术仍存在基因组覆盖不均、PCR错误、等位基因脱落、对高GC含量区域的偏向性、低可映射率(约20%至30%)以及受限的捕获率(约40%)等局限性。这些技术偏差不仅降低了数据的质量和可靠性,还增加了后续生物信息学分析的复杂性,需要专门的算法来估算缺失信息并校正错误,从而限制了这些技术在更广泛研究群体中的普及性。尽管如此,单细胞方法和计算分析的持续进展已开始通过估算缺失信息来克服这些挑战。
此外,不同实验批次、测序平台和测量方法之间的差异导致了显著的批次效应和数据异质性,使得来自不同来源的单细胞数据难以有效整合和比较。尽管实验方法在同时分析多种组学模态方面取得了进展,但不同组学层包含不同的特征空间,使得多组学数据整合成为一大障碍。因此,开发能够有效整合不同模态数据的计算方法至关重要,以实现更全面的生物学见解。
应对这些挑战,需要开发和应用先进的生物信息学工具和计算算法,尤其是在数据标准化、多模态数据对齐、轨迹推断和细胞间通讯分析等方面。当前计算分析领域正经历快速发展,旨在处理高数据维度、高噪音以及数据稀疏性问题,但要实现完全成熟和标准化仍需持续努力。未来的研究应着力于开发更鲁棒、更具解释性和可扩展性的算法,例如:1) 针对稀疏性问题,开发基于深度学习的生成模型或自适应插补算法,以更准确地恢复缺失信息并消除噪声;2) 针对多模态整合,探索跨模态图神经网络或张量分解方法,以捕捉更复杂的特征关联和多层级调控网络;3) 针对批次效应,开发能够自适应不同批次特征的联邦学习或迁移学习方法,从而提高数据整合后的泛化能力和可比性。同时,结合新兴的空间组学技术,以弥补传统单细胞测序中空间信息丢失的不足,将是进一步揭示肿瘤免疫微环境复杂性的重要途径。
4.1 数据预处理与质控
单细胞多组学数据分析面临诸多挑战,其中包括数据稀疏性、高维度、PCR错误、等位基因丢失以及低覆盖率导致的极端数据稀疏性,这些因素严重影响了下游分析的准确性与可靠性。此外,前端组织解离过程不可避免地破坏了生物样本的空间结构,从而消除了有助于细胞生物学身份的关键信息层,这也为数据分析带来了额外的复杂性。因此,有效的数据预处理和质量控制是单细胞多组学数据分析流程中至关重要的起始步骤,旨在识别并去除低质量细胞、处理批次效应,以及通过估算缺失信息来克服数据稀疏性和不均一覆盖的问题,从而确保数据的可靠性和可比性。
在数据预处理阶段,关键步骤包括去噪、标准化和批次校正。去噪处理旨在减少技术噪音对数据的影响,提高信号的准确性。标准化则确保不同细胞或样本间的数据具有可比性,消除由于测序深度或文库大小差异带来的系统偏差。批次效应是单细胞数据分析中普遍存在的问题,它由实验操作批次差异引起,而非生物学差异。为了解决这一问题,研究人员已开发出多种计算方法进行批次校正,以整合来自不同实验批次的数据,确保其可靠性和可比性。例如,Seurat v4通过加权最近邻(WNN)分析整合多种模态的单细胞测量结果,并共同定义细胞状态。这种无监督策略能够学习细胞特异性模态的“权重”,以确定其在后续分析步骤中的重要性,从而有效应对数据稀疏性和高维度挑战。
尽管计算工具和技术在快速发展以应对单细胞数据分析中的高数据维度、高噪音和数据稀疏性等挑战,但该领域仍需进一步努力才能实现完全成熟和标准化。未来的研究方向应着重于开发更稳健的计算方法,以更有效地处理数据稀疏性,估算缺失信息,并克服样本量限制和方法论约束。此外,结合新兴的空间组学技术,以弥补传统单细胞测序中空间信息丢失的不足,将是进一步揭示肿瘤免疫微环境复杂性的重要途径。
4.2 细胞聚类与亚群识别
单细胞技术,特别是单细胞RNA测序(scRNA-seq)和质谱流式细胞术(CyTOF),产生了高维度、稀疏的数据集,为深入理解肿瘤免疫微环境(TME)中的复杂细胞亚群提供了前所未有的机会。这些数据集的有效分析依赖于一系列计算方法,旨在从异质性数据中识别并定义不同的细胞亚群。
在单细胞数据分析中,降维是关键的第一步,它有助于将高维数据映射到低维空间,从而更有效地进行可视化和聚类。常用的降维技术包括均匀流形逼近和投影(UMAP)和t-分布随机邻域嵌入(t-SNE)。这些方法能够揭示数据中的内在结构,使细胞群集在低维空间中得以区分。随后,诸如Louvain和Leiden等聚类算法被广泛应用于识别细胞亚群。这些算法通过迭代优化,将相似的细胞归为一类,从而实现对细胞类型的无监督聚类和分类,甚至识别稀有细胞亚群。
除了静态的细胞亚群识别,描绘细胞发育谱系和动力学对于理解TME中的细胞状态转换至关重要。轨迹推断方法通过伪时间分析和RNA Velocity等技术,可以预测细胞分化轨迹,深入理解细胞分化和状态转换过程。Monocle/Monocle 2是其中一类广为应用的算法,它能够基于单细胞转录组数据推断细胞的伪时间排序,从而揭示细胞从一个状态向另一个状态演变的过程。
然而,这些轨迹推断方法存在一定的局限性。Saelens等人对45种轨迹推断方法进行了基准测试,结果表明计算方法的预设轨迹拓扑结构会影响推断结果,且不同方法在不同数据集上的表现存在差异。这些方法的细胞轨迹估计主要通过细胞-细胞距离计算,本质上是表型上的预测,可能不一定反映细胞谱系之间的真实遗传关系。
为了解决这一局限性,一些新方法被开发出来。RNA Velocity通过量化单细胞中剪接和未剪接转录本的丰度,推断细胞在短时间内的转录状态变化方向,并将具有相似转录本剪接状态的细胞连接起来,从而提供更接近真实细胞状态变化的动态信息。另一个名为CytoTRACE的方法,利用检测到的基因数量来反映单细胞的发育潜力,为描绘细胞轨迹提供了稳健的性能。尽管这些方法提供了强大的分析能力,但scRNA-seq衍生的细胞图谱仍然缺少细胞身份的几个关键方面,包括表观遗传状态、蛋白质谱和细胞的空间位置信息。
未来的研究方向应着重于开发能够整合多组学数据(如基因组、表观基因组、蛋白质组和空间组学数据)的计算方法,以提供更全面、更准确的细胞身份和轨迹信息。此外,针对不同数据集特性,优化和开发更具鲁棒性的轨迹推断算法,以克服当前方法的局限性,从而更精确地描绘TME中的细胞动态变化和相互作用。
4.3 多组学数据整合与分析
单细胞多组学数据整合分析是全面理解细胞生物学,特别是肿瘤免疫微环境(TME)异质性和组织结构的关键。然而,这一过程面临诸多计算挑战,包括高数据维度、稀疏性、低映射率、受限的捕获率、高水平的PCR重复序列以及来自不同实验或平台的批次效应等。为了克服这些挑战,研究人员开发了多种计算策略和工具,旨在将不同组学数据投影到共同的特征空间或非线性流形中,以揭示细胞间的相似性,从而实现更精确的细胞类型描绘和功能定义。
当前整合方法的核心在于应对不同组学数据特征差异和批次效应。一种常用的策略是共享最近邻图(SNN),通过在共嵌入空间中寻找互最近邻细胞对作为“锚点”来指导数据集整合。例如,Seurat v3和LIGER等工具利用典型相关分析(CCA)或非负矩阵分解(NMF)来识别共享的生物学空间。另一种方法是估计单个组学内部的细胞距离,然后计算“加权最近邻”距离进行多组学数据的整合分析。此外,多视图机器学习、深度生成模型以及基于修改统计框架识别跨数据模态低维变异的方法也被广泛应用于多组学单细胞数据整合。近年来,随着真实多组学分析技术的进步,如GLUE、StabMap、Cobolt、MultiVI以及Seurat v5中的“Bridge Integration”等方法,通过利用配对的多组学配置文件,显著提高了非配对数据集的整合准确性。
多组学数据整合的目的是在更高维度的特征空间中,基于细胞相似性来描绘细胞类型,进而深入理解复杂的生物学过程。例如,整合单细胞RNA测序(scRNA-seq)和单细胞ATAC测序(scATAC-seq)数据可以更有效地揭示转录调控机制,而结合蛋白质组数据则能更精确地定义细胞表型。此外,将基因组、表观基因组、转录组、蛋白质组和空间组织等多模态数据集整合,能够扩展单细胞基因组学在免疫学研究中的能力。这种整合有助于实现直接从人类样本进行回顾性谱系追踪,从而理解免疫细胞在TME刺激下的分化、激活和耗竭机制。同时,整合表观基因组、转录组和蛋白质组有助于更准确地定义细胞身份,全面阐明细胞的功能可塑性、历史和未来潜能。加入空间定位信息则进一步完善了细胞身份的定义,这对于理解免疫系统细胞之间的相互作用以及肿瘤与其他TME细胞类型之间的串扰至关重要。
在多组学数据整合领域,目前的主流研究强调以数据驱动的计算方法来解决挑战,例如Seurat和LIGER等工具的广泛应用,但新兴研究已开始质疑其在捕捉复杂生物学机制方面的长期有效性。当前整合方法的共识在于利用计算模型识别不同组学数据间的共同变异,并消除批次效应;分歧则在于如何最优地加权不同模态的信息,以及如何处理非线性关系和不同数据类型间的异质性。例如,系统生物学方法,利用数学模型来提供对TME所有组成部分(包括细胞类型及其细胞内和细胞间调控机制)的整体视图,被认为是应对多模态数据整合挑战的关键策略。这些模型不仅能够揭示肿瘤发生的机制,进行实验验证的预测,还可用于描述细胞内信号传导以理解耐药性并提出个性化治疗方案。
展望未来,随着空间分辨RNA和蛋白质表达数据分析技术的发展,迫切需要新的计算方法来研究考虑空间信息的细胞内网络。未来研究方向将从主要以数据驱动为主的方法,扩展到包含细胞类型特异性细胞内通路和配体-受体相互作用的先验知识的机制模型,这将有助于更全面地理解细胞间相互作用和肿瘤的发生发展,并为个性化治疗提供新的策略。
4.4 细胞间通讯推断
肿瘤免疫微环境(TME)中细胞间的复杂相互作用是理解肿瘤进展、免疫逃逸及治疗响应的关键。单细胞测序技术的发展为在单细胞分辨率下解析这些相互作用提供了前所未有的机会。在单细胞多组学数据的基础上,计算方法已成为推断TME中细胞间通讯网络的重要工具,其核心在于利用配体-受体数据库与网络分析工具来识别关键的信号通路。
推断细胞间通讯网络通常涉及以下几个关键步骤。首先,需要构建全面的配体-受体数据库,这些数据库整合了已知或预测的细胞表面配体及其对应的受体,以及分泌型分子与其靶细胞受体之间的相互作用。这些数据库是进行通讯推断的生物学基础,确保了推断结果的生物学合理性。其次,基于单细胞数据(如单细胞RNA测序数据)量化不同细胞类型中配体和受体的表达水平。通过比较细胞群体中配体和受体的共表达模式,可以初步筛选出潜在的相互作用对。第三,利用网络分析工具进一步解析细胞间的通讯模式。这些工具可以将细胞视为节点,将推断出的配体-受体相互作用视为边,构建出细胞间通讯网络。通过分析网络的拓扑结构,可以识别出在TME中发挥主导作用的关键信号通路和“中心”细胞类型。例如,可以计算节点的度(degree centrality)来识别与众多其他细胞类型相互作用的细胞,或计算介数中心性(betweenness centrality)来识别在信息流中起桥梁作用的细胞。
目前,多种计算方法已被开发用于单细胞数据的细胞间通讯推断。这些方法通常结合了统计学模型、机器学习算法和生物信息学工具,以提高推断的准确性和鲁棒性。然而,该领域仍面临诸多挑战,例如,现有配体-受体数据库的完整性与准确性仍有待提高;同时,如何区分直接相互作用与间接影响,以及如何整合多组学数据(如蛋白质组学和空间转录组学数据)以更全面地刻画细胞间通讯,是未来研究需要解决的关键问题。未来的研究方向将侧重于开发更为精细的计算模型,结合时间序列数据分析动态的细胞间通讯变化,并利用人工智能技术从大规模单细胞数据中挖掘未知的通讯模式,从而更深入地揭示TME的复杂性,为肿瘤的精准治疗提供新的靶点和策略。
4.5 单细胞谱系追踪
单细胞谱系追踪技术在解析肿瘤免疫微环境(TME)中免疫细胞的分化、激活、耗竭及记忆形成机制方面展现出巨大潜力,尤其是在人体样本研究中。通过将细胞的遗传谱系与其功能表型相联系,谱系追踪能够揭示细胞演进的真实路径,从而补充并深化伪时间分析(pseudotime analysis)所提供的见解。
目前,单细胞谱系追踪主要分为前瞻性(prospective)和回顾性(retrospective)两种方法。前瞻性追踪方法通常通过遗传操作引入独特的DNA条形码来标记细胞,从而精确追踪不同细胞谱系的克隆动态。然而,此类方法操作复杂,且目前主要适用于体外实验和动物模型系统,在人类样本中应用受限。
相比之下,回顾性谱系追踪方法利用细胞发育过程中自然发生的体细胞突变作为内源性标记,已广泛应用于人类样本分析,有效克服了前瞻性方法的局限性。常用的回顾性标记包括T细胞受体(TCR)/B细胞受体(BCR)序列、拷贝数变异(CNVs)、单核苷酸变异(SNVs)、逆转录转座子(如LINE-1)以及线粒体DNA(mtDNA)突变。TCR序列作为高多态性遗传标记,结合单细胞转录组分析,已为T细胞分化过程提供了重要见解,能够将T细胞的遗传谱系与其转录组定义的功能表型联系起来。在人类TME背景下,TCR克隆型分析揭示了共享TCR克隆型可以跨越广泛的T细胞分化状态,表明这些细胞状态在发育上是相互关联的;而某些TCR克隆型则几乎仅限于特定的细胞亚群,暗示其独特的发育谱系。此外,基于mtDNA突变的谱系追踪已成功应用于研究人类肝癌TME中巨噬细胞的谱系,其结果与基于RNA velocity的预测结果一致,进一步验证了回顾性追踪方法的可靠性。
尽管谱系追踪技术取得了显著进展,但在人类TME中免疫细胞发育谱系的直接了解仍然非常有限,存在诸多未解决的关键问题。例如,肿瘤内调节性T细胞(Treg细胞)的起源仍不明确,因为其大部分TCR克隆型是其独有的,仅有一小部分与血液或邻近正常组织中的Treg细胞或肿瘤内辅助性T细胞(Th细胞)共享。另一个挑战是程序性死亡受体-1(PD-1)阻断后扩增的T细胞群体的细胞起源问题。例如,在皮肤癌患者中观察到的“克隆型替代”现象,即PD-1阻断后扩增的克隆型与预处理TME中存在的克隆型不同,这可能表明TME中大多数预先存在的T细胞的再激活能力有限。然而,这些扩增的T细胞克隆是源自TME外新启动的T细胞,还是源自先前因稀有而未检测到的“祖细胞样”T细胞的克隆扩增和分化,仍有待深入研究。未来的研究应致力于开发更精确、更广泛适用于人类样本的谱系追踪技术,并结合多组学数据,以期全面解析肿瘤免疫细胞的动态演进过程。
5. 结论与展望
单细胞多组学技术作为新兴的生物学研究范式,极大地推动了我们对肿瘤免疫微环境(TME)复杂性的理解。通过深入解析肿瘤免疫微环境中细胞的高度异质性、动态性和可塑性以及复杂的细胞间相互作用,单细胞多组学超越了传统批量组学分析的局限性,为癌症精准医疗开辟了新的途径。该技术不仅揭示了肿瘤发生、免疫逃逸及治疗响应的深层机制,还为预测生物标志物识别、稀有细胞群描绘以及治疗耐药机制的阐明提供了强有力的数据支持。
尽管单细胞多组学技术潜力巨大,但当前其广泛应用和临床转化仍面临多重挑战。核心瓶颈包括:技术通量受限与成本高昂,制约了大规模患者队列研究的开展和发现的普适性;数据质量受损,表现为捕获效率低、数据稀疏性高以及存在聚合酶链式反应(PCR)错误和等位基因丢失等问题;以及传统技术在组织解离过程中空间信息丢失,无法全面描绘细胞的原位互作网络。此外,数据分析的复杂性,如批次效应和多模态数据整合的挑战,以及在临床转化过程中,标准化方法缺失、伦理考量和个体化差异带来的“翻译”鸿沟,都亟待解决。
未来的研究和发展将聚焦于克服这些挑战,以期实现单细胞多组学在肿瘤免疫微环境研究和精准医疗中的全面应用。关键方向包括:推动更高通量、更低成本的新技术发展,如组合索引和空间多组学技术,以提升数据质量和空间分辨率;开发更先进的计算生物学和人工智能算法,以有效整合和解析多模态、高维度、稀疏数据,并从数据中提取深层生物学机制;加强研究与临床之间的转化,建立大规模多中心研究框架,标准化数据采集与分析流程,并着重功能验证,以确保生物标志物的稳定性和可重复性,从而指导个性化癌症治疗方案的制定。通过系统性地解决这些问题,单细胞多组学有望最终实现对肿瘤表型的精准操控,克服免疫抑制,恢复免疫监视,并显著提升癌症患者的治疗效果和预后。
5.1 新技术与方法的发展
单细胞多组学技术的持续创新极大地拓展了肿瘤免疫微环境(TME)研究的深度与广度。其中,空间多组学技术的发展尤为关键,它弥补了传统单细胞分离技术在组织解离过程中丢失空间信息的局限性,从而能够提供细胞在组织中的原位分布及其相互作用的详细信息。
当前,多种空间多组学技术正在逐步成熟。成像技术在肿瘤组织切片上的应用,如免疫组织化学(IHC)和免疫荧光(IF),已成为蛋白质表型分析的成熟手段,它们能够以亚细胞分辨率检测基因和蛋白质的表达,并保留细胞的空间位置。尽管多重IHC结合细胞分割和标记物分类软件可描绘TME的细胞结构,但其标记物数量有限,限制了对复杂TME的全面解析。为克服这一限制,新兴的基于抗体的成像技术,如成像质谱流式细胞术(IMC)和多重离子束成像(MIBI),通过量化多达40种标记物,能够生成类似组学的数据,显著提升了分子信息的全面性。这些技术不仅能够提供肿瘤、免疫和基质细胞、血管和细胞外基质(ECM)的空间定位,还能以亚细胞分辨率进行分子检测。此外,IMC的最新进展实现了同一细胞上转录组和蛋白质组标记物的同步测量,揭示了如HER2 mRNA和蛋白质表达在单细胞水平上可能存在的非一致性,并强调了分析空间模式对于理解组织生物学的重要性,例如CXCL10表达细胞簇集形成的组织基序在肿瘤浸润性免疫细胞研究中的发现。
空间转录组学作为另一个重要分支,其方法可分为荧光类和测序类。例如,seqFISH+能够以亚细胞分辨率对组织中超过10,000个转录本进行超分辨成像,但其复杂的实验程序和探针设计仍是应用挑战。基于scRNA-seq的细胞条形码策略可实现组织中局部细胞的原位条形码,但将物理位置与条形码序列解复用是其主要挑战。Slide-seq等空间测序技术的开发,旨在弥补现有单细胞技术在组织解离过程中丢失空间信息的缺点,并有望在不久的将来以单细胞分辨率解析重要的细胞相互作用。
除了空间多组学技术,组合索引技术的发展显著提升了单细胞测序的可扩展性和通量,使其通量接近传统流式细胞术(FACS)和质谱流式细胞术(CyToF)的水平。体内单细胞分析技术也正在发展中,以克服现有局限性。长读长测序技术虽然有望推进细胞遗传变异的检测,但仍面临测序准确性低以及从临床样本中获取完整DNA和全长RNA的困难。
未来单细胞组学技术将进一步聚焦于克服现有局限性,并有望通过整合多组学技术对单细胞进行更深入的分析,以全面解析复杂的肿瘤生态系统。
5.2 计算生物学、人工智能与大数据分析的融合
单细胞多组学数据分析面临高维度、稀疏性、批次效应以及多模态数据整合等挑战,这凸显了计算生物学、人工智能(AI)与大数据分析融合的重要性。人工智能和机器学习(ML)技术在处理复杂单细胞多组学数据方面展现出巨大潜力,可用于自动化细胞类型识别、疾病状态预测和药物响应生物标志物发现,并加速分析流程和促进生物学解释。
在单细胞数据模式识别方面,大数据分析与深度学习模型能够有效地挖掘数据深层信息。例如,SCENIC+、FigR和MIRA等方法通过建模配对的转录组和单细胞染色质开放性测序(scATAC-seq)的联合变异,从而关联增强子与基因,并识别驱动细胞命运决定的转录因子。在预测模型构建方面,MultiVelo通过整合染色质可及性和基因表达信息,能够预测未来的细胞状态,包括染色质可及性转换和基因剪接状态的动态变化。此外,CellOracle利用多组学数据学习基因调控网络,进而推断体外扰动的结果,这对于理解细胞行为和疾病机制具有重要意义。
计算系统模型在肿瘤免疫微环境(TME)的理解与精准免疫肿瘤学中扮演着关键角色。这些模型能够整合单细胞和批量多组学数据,有助于理解TME组分的运作机制,并预测治疗对整个系统的影响。数学模型已被应用于免疫疗法和联合治疗的计算机筛选,从而为个性化治疗方案的制定提供依据。动态模型被用于描述细胞内信号传导以理解耐药性并提出个性化疗法,且可进一步扩展至研究细胞外信号传导。细胞间相互作用可以通过将单个细胞视为代理或黑箱进行建模,进而理解细胞相互作用、体外测试不同治疗干预的效果以及研究肿瘤的起始和进展。
在克服小样本队列和数据异质性带来的挑战方面,计算分析的进步已开始通过信息插补(imputing missing information)等方法弥补单细胞多组学数据中因基因组覆盖不均、高GC偏好、低映射率、捕获率受限以及高PCR重复序列等问题所导致的数据局限性。未来,随着空间分辨RNA和蛋白质表达数据分析技术的发展,迫切需要新的计算方法来研究考虑空间信息的亚细胞标记物。这些方法将从主要的数据驱动方法扩展到整合细胞类型特异性细胞内通路和配体-受体相互作用先验知识的机制模型。新的计算工具对于提高整合数据质量、促进生物学解释和加速分析过程具有重要价值,其开发是未来研究的重要方向。
5.3 临床转化与精准医疗
单细胞多组学技术作为新兴的生物学工具,正逐步从基础研究领域向临床转化迈进,为肿瘤精准医疗开辟了新的视角与途径。肿瘤的复杂性,尤其是肿瘤免疫微环境(TME)的高度异质性和动态性,使得传统的批量组学分析难以全面捕捉其全貌,并精确预测治疗反应。单细胞多组学技术通过揭示肿瘤内部的异质性、免疫编辑和免疫原性等关键机制,克服了传统方法的局限,从而为个性化免疫治疗方案的制定提供了前所未有的深入见解。
在指导个性化免疫治疗方面,单细胞多组学数据展现出巨大潜力。通过整合不同组学数据,研究人员能够阐明肿瘤细胞表型在肿瘤进展和治疗过程中的演变,进而优化CAR-T疗法、识别新的治疗靶点,并监测循环肿瘤细胞。例如,新抗原的精准表征为个性化疫苗接种策略提供了新的方向,尽管其临床转化仍需在精确度和时间效率方面进一步优化。此外,单细胞多组学有助于识别癌症预后和预测治疗效果的分子生物标志物,例如肿瘤突变负荷、微卫星不稳定性、PD-L1表达等,尽管目前尚无单一标志物能够明确区分免疫检查点阻断剂(ICBs)的响应者和非响应者。未来的研究,如通过纵向样本的多组学数据分析,有望发现更具信息量的预测性生物标志物,从而实现更有效、毒性更小的精准免疫治疗。
功能筛选作为精准医疗的补充方法,能够直接检测肿瘤对药物扰动的反应。利用小鼠模型和肿瘤类器官进行体外药物测试,可以大规模筛选药物扰动,特别是结合类器官与免疫细胞共培养技术,有望实现免疫检查点阻断剂的功能筛选和用于免疫治疗的肿瘤反应性T细胞的选择。此外,微流控技术的应用使得直接在肿瘤活检组织上进行药物筛选成为可能,并可进一步调整以识别免疫检查点阻断剂的协同伙伴,加速新治疗靶点的发现和优化免疫治疗策略。
在推动临床转化方面,人类肿瘤图谱网络(HTAN)等大型项目发挥着至关重要的作用。HTAN项目提出了肿瘤演进过程中三维细胞图谱的新概念,旨在通过整合人类肿瘤的分子、空间和临床检查数据,揭示肿瘤发生的基本机制,并发现用于癌症筛查、肿瘤转移、癌症免疫治疗和药物反应的新生物标志物。尽管当前单细胞方法的成本较高导致纳入的患者队列较小,可能造成生物学发现的不一致和不可重复性,但通过整合大规模跨平台、跨组学和跨物种的数据,并结合相关功能和临床信息,最终将深化对人类抗肿瘤免疫的理解,进而改进患者分层、生物标志物发现和可药物靶点识别,实现癌症患者的精准医疗。对TME作为多细胞系统的全面理解对于合理设计临床试验和提供预测性、动态生物标志物至关重要。
5.4 未解决的问题与未来研究方向
尽管单细胞多组学技术在解析肿瘤免疫微环境(TME)方面取得了显著进展,但仍面临诸多未解决的关键问题与挑战,亟待未来研究予以克服 。
一、技术瓶颈与优化方向
当前的单细胞技术仍存在一系列技术局限性。首先,在数据获取层面,普遍存在捕获率受限、PCR错误、等位基因丢失、基因组覆盖不均以及高GC偏好等问题,这些因素直接影响了数据质量与完整性,并导致较低的映射率(20%至30%)和捕获率(约40%) 。其次,现有技术在通量和成本方面仍存在瓶颈,高昂的成本限制了大规模患者队列研究的开展,使得生物学发现可能不一致且难以重复,迫切需要更多的外部验证 。
针对这些技术局限性,未来的研究方向应包括:
- 提高通量与降低成本:开发更具成本效益的高通量单细胞测序平台,以支持更大规模的临床研究,从而提高发现的可重复性和可靠性 。
- 改善数据质量:优化单细胞分离、建库和测序流程,减少技术假象,提高数据完整性,例如针对流式细胞术时间飞行质谱(CyTOF)和单细胞RNA测序(scRNA-seq)的局限性,提出增加可检测通道数量、优化scRNA-seq系统以生成高质量统一数据 。
- 整合多模态数据:当前单细胞转录组数据纯粹的表型预测可能不反映真实的细胞谱系关系,且缺乏表观遗传状态、蛋白质谱和空间位置等关键信息 。因此,未来的研究应着重于将基因组、表观基因组、转录组、蛋白质组和空间组织等多模态数据整合,从而更全面地解析TME的复杂性,并实现直接从人类样本进行回顾性谱系追踪 。技术创新如空间转录组学和组合索引技术有望弥补现有局限,实现更高通量和空间分辨率的分析,为精准免疫治疗的开发提供新思路 。
二、数据分析与整合挑战
单细胞多组学数据分析面临的挑战主要包括数据稀疏性、批次效应以及现有整合算法的局限性 。尽管计算方法已开始通过信息插补等方式克服数据缺失问题,但仍需要进一步的功能验证来确认这些发现的生物学意义 。
未来的研究方向应包括:
- 开发新型计算工具:需要开发新的计算工具以提高数据整合质量、促进生物学解释和加快分析流程,特别是在系统整合多组学数据以揭示TME中导致患者差异反应的特征方面,这需要更精细的计算方法和生物信息学工具 。
- 推动生物信息学与系统生物学融合:生物信息学和系统生物学将在提取新的机制性见解和指导免疫肿瘤学领域中发挥核心作用,通过系统生物学和计算系统模型整合单细胞和批量多组学数据,理解TME组分如何运作,并预测疗法对整个系统的影响 。
三、临床转化中的问题与前景
单细胞多组学技术在临床转化中面临大规模多中心研究的标准化、不同技术平台数据的可比性、数据共享和伦理问题等关键挑战 。不同临床研究中组织位点、样本状态、分离方法和样本切除时间点的差异,导致临床领域发现的单细胞生物标志物不稳定且不可重复 。此外,仍需对现有发现进行功能验证 。
未来的研究应专注于:
- 建立大规模临床研究框架:迫切需要一个更可行的单细胞框架来执行大规模临床研究,并建立用于共享和利用已发表癌症领域单细胞数据的资源,以克服小样本队列和生物标志物不稳定的问题,从而促进未来的临床转化 。
- 加强功能验证:尽管单细胞多组学实验提供了诱人的证据,但仍需要进一步的功能验证来确认这些结果的生物学意义和临床转化价值 。例如,发展功能筛选技术,利用小鼠模型、肿瘤类器官和微流控技术进行体外药物筛选,以直接评估肿瘤对药物的反应,从而为个性化治疗提供依据 。
- 识别预测性生物标志物:在癌症免疫治疗中,迫切需要识别预测性生物标志物,这要求在数据可用性和算法优化方面进行深入研究,尤其是在组合疗法方面,以推动免疫检查点阻断剂(ICBs)与靶向药物的合理组合,实现协同作用,将免疫疗法的长期临床益处扩展到更大部分的癌症患者 。
最终,通过克服上述挑战,单细胞多组学方法将指导癌症中单细胞特征的检测、免疫操作以克服免疫抑制、恢复免疫监视,并最终稳定或逆转癌症患者的肿瘤表型,实现更精准和个性化的癌症治疗 。
References
Single-Cell Sequencing in Cancer: Recent Applications to Immunogenomics and Multi-omics Tools https://pmc.ncbi.nlm.nih.gov/articles/PMC6440661/
Applications of Single-Cell Omics to Dissect Tumor Microenvironment - Frontiers https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2020.548719/full
Multi-Omics Profiling of the Tumor Microenvironment: Paving the Way to Precision Immuno-Oncology - Frontiers https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2018.00430/pdf
The technological landscape and applications of single-cell multi-omics - PubMed Central https://pmc.ncbi.nlm.nih.gov/articles/PMC10242609/
Applications of Single-Cell Omics in Tumor Immunology - Frontiers https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2021.697412/full